
Sindromul cri du chat
Date generale. Sindromul “cri du chat” este o boală rară, determinată de monosomia parţială 5p. Incidenţa bolii este 1/50000 de naşteri.
Genetică. Majoritatea semnelor clinice sunt determinate de deleţia (absenţa) unei mici regiuni cromosomice, localizate în regiunea 5p15.2, la capătul terminal al braţului scurt al cromosomului 5. Semnele şi simptomele bolii sunt determinate de pierderea mai multor gene localizate pe fragmentul cromosomic care lipseşte.
Diagnostic. La sugar semnele clinice sunt: plâns caracteristic, similar mieunatului unui pisoi, microcefalie (perimetru cranian mai mic decât normal), hipotonie musculară (tonus muscular scăzut), dismorfie cranio-facială cu facies rotund, hipertelorism (distanţă interpupilară mărită), urechi jos inserate. La copilul mare şi adult dismorfia facială include faciesul îngust, micrognaţia (maxilar inferior scurt) şi ştergerea unghiului mandibular. Dezvoltarea intelectuală este deseori întârziată şi de multe ori există malformaţii cardiace (defect septal atrial, defect septal ventricular, persistenţa canalului arterial) sau genito-urinare (rinichi în potcoavă, ectopie renală, hidronefroză).
Diagnosticul este confirmat de efectuarea de analize genetice.
Analiza cromosomică clasică permite diagnosticul doar în circa 10% din cazuri, când deleţia este suficient de mare, dar pentru aceasta este nevoie de obţinerea de comosomi suficient de lungi (cu o rezoluţie de minimum 5,5 Mb).
La cazurile sugestive clinic la care analiza cromosomică clasică nu indică existenţa unei deleţii, este necesară efectuarea unui test FISH (hibridare fluorescentă in situ) folosind sonde specifice regiunii 5p15 care permite identificarea de deleţii mici, inframicroscopice, caracterizate prin absenţa unui fragment din cromosomul 5, imposibil de pus în evidenţă prin analiza cromosomică clasică.
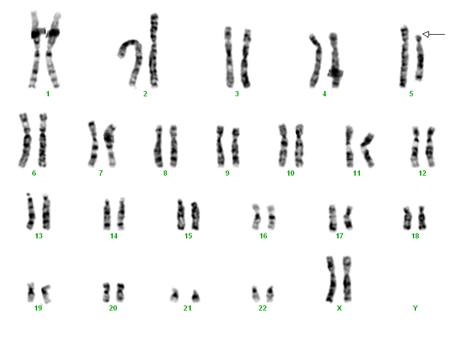
Deletie 5p
În 85% din cazurile de sindrom cri du chat, deleţia 5p este omogenă (prezentă în toate celulele) şi de novo (rezultată printr-o mutaţie nouă apărută în cursul formării gameţilor la unul dintre părinţii sau în primele etape ale dezvoltării embrionare). În restul cazurilor deleţia este secundară prezenţei la unul dintre părinţi a unei anomalii structurale echilibrate (translocaţie sau inserţie).
Tratament. Nu există terapie specifică care să ducă la vindecarea bolii, ci doar tratamente care ameliorează anumite manifestări ale acesteia. Pacienţii cu sindrom cri-du-chat necesită colaborarea dintre părinţi şi medici, cu scopul stimulării fizice şi intelectuale, pentru a-şi putea atinge potenţialul maxim. Este util tratamentul logopedic pentru dezvoltarea limbajului şi kinetoterapia pentru ameliorarea tonusului muscular. În cazul existenţei de anomalii viscerale severe, se impune corecţia chirurgicală a anomaliilor congenitale respective.
Prognosticul. La pacienţii fără anomalii cardiace, speranţa de viaţă nu este major modificată, bolnavii prezentând un deficit de dezvoltare. Evoluția depinde de gravitatea dizabilităţii intelectuale şi existenţa unor anomalii fizice. Mai mult de jumătate dintre copii dezvoltă limbajul verbal şi comunică cu anturajul. În restul cazurilor, dezvoltarea limbajului verbal este limitată la propoziţii scurte, însoţite de gesturi (comunicare non-verbală). Unii pacienţi pot avea: dificultăţi de alimentare, hiperactivitate, deformări ale coloanei vertebrale (scolioză). Un număr mic de copii se nasc cu malformaţii ale organelor interne care le pot pune viaţa în pericol. În general, pacienţii au o atitudine prietenoasă, sunt veseli şi se pot integra în mediul familial. În cazul, unei dizabilităţi mintale severe ei nu se pot îngriji singuri şi nu pot avea o viaţă socială normală.
Profilaxie. La părinţii tuturor copiilor cu sindrom cri du chat este necesară efectuarea cariotipului. Dacă cei doi părinţi sunt indemni, riscul de a avea un alt copil afectat este nesemnificativ. În schimb, dacă există o translocaţie echilibrată riscul de recurenţă este de aproximativ 10%., astfel încât se impune efectuarea unui diagnostic prenatal prin tehnica FISH, folosind sonde corespunzătoare regiunii 5p15 .
Diagnostic prenatal. Depistarea prenatală a sindromului cri du chat la cuplurile fără istoric familial pozitiv este întâmplătoare, analiza cromosomică prenatală fiind impusă de alte motivaţii, precum dublu test sau triplu test cu valori de alarmă pentru alte boli cromosomice. Existenţa unor modificări ecografice cardiace, poate sugera existenţa bolii, dar de regulă sunt căutate anomalii cromosomice caracteristice unor boli cromosomice mai frecvente, precum sindromul Down (trisomia 21) sau sindromul Turner (monosomia X).
Prof.dr. Eusebiu Vlad Gorduza